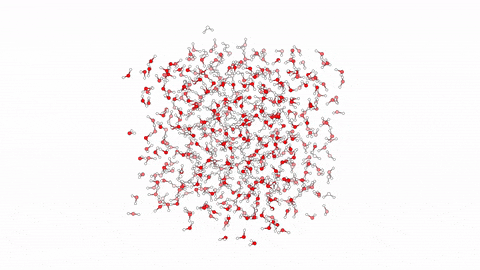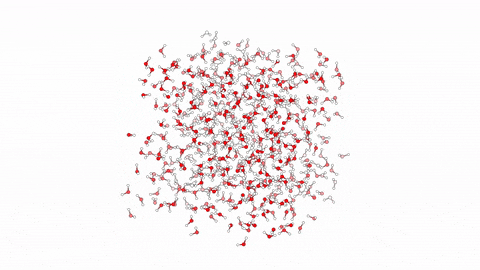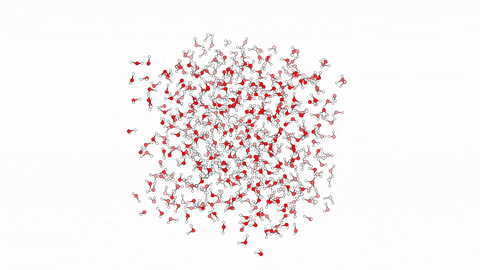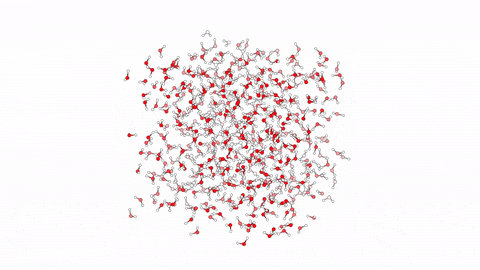
Molecular dynamics (MD) is a powerful computational tool used to model biological processes and explore their mechanisms at the atomic/molecular level. This technique has enabled us to investigate atom-to-atom interactions of various antibacterial agents ranging from small molecules to branched peptides.
While atomistic simulations provide valuable insights, limitations related to computational cost, accuracy, and accessible simulation timescales must always be considered. Many key molecular interactions occur over timescales that standard MD simulations often struggle to capture.
To address these challenges, enhanced sampling techniques have become essential tools in studying complex biological phenomena. Various enhanced sampling strategies, such as metadynamics, have been developed to address the limitations of conventional molecular simulations, which often struggle to capture rare events and slow transitions. These approaches accelerate the exploration of molecular landscapes, enabling more efficient estimation of thermodynamic properties and dynamic behaviors. By facilitating access to otherwise inaccessible regions of the system’s configuration space, enhanced sampling techniques have become essential in studying complex biological processes, providing valuable insights into their structural and functional mechanisms.
In this video, we showcase a simulation of 10 linear antibacterial peptides in solution, performed over 10ns using enhanced sampling. The method focuses on hydration and dehydration of peptides, leading to capture the early stages of peptide aggregation, a phenomenon crucial in understanding their biological behavior.
This blog was written by Saha Satvati, one of the PhD candidates working in the SSBB consortium SSBB. Saha is pursuing his PhD trajectory at the Istituto Italiano di Tecnologia and SetLance.
